Leaving group
In chemistry, a leaving group is a molecular fragment that departs with a pair of electrons in heterolytic bond cleavage. Leaving groups can be anions or neutral molecules, but in either case it is crucial that the leaving group be able to stabilize the additional electron density that results from bond heterolysis. Common anionic leaving groups are halides such as Cl−, Br−, and I−, and sulfonate esters such as tosylate (TsO−). Fluoride (F−) functions as a leaving group in the nerve-agent sarin gas. Common neutral molecule leaving groups are water and ammonia. Leaving groups may also be positively charged cations (such as H+ released during the nitration of benzene); these are also known specifically as electrofuges.[1][2]
Contents
1 Leaving group ability
2 Contextual differences in leaving group ability
3 Activation
4 In conjugate base eliminations
5 "Super" and "Hyper" leaving groups
6 See also
7 References
Leaving group ability
The physical manifestation of leaving group ability is the rate at which a reaction takes place. Good leaving groups give fast reactions. By transition state theory, this implies that reactions involving good leaving groups have low activation barriers leading to relatively stable transition states.
It is helpful to consider the concept of leaving group ability in the case of the first step of an SN1/E1 reaction with an anionic leaving group (ionization), while keeping in mind that this concept can be generalized to all reactions that involve leaving groups. Because the leaving group bears a larger negative charge in the transition state (and products) than in the starting material, a good leaving group must be able to stabilize this negative charge, i.e. form stable anions. A good measure of anion stability is the pKa of an anion's conjugate acid, and leaving group ability indeed generally follows this trend, with a lower pKaH being associated with better leaving group ability.

In an ionization reaction, as in all reactions that involve leaving group departure, the leaving group bears a larger negative charge in the transition state and products than it does in the starting materials
The correlation between pKaH and leaving group ability, however, is not perfect. Leaving group ability represents the difference in energy between starting materials and a transition state (ΔG‡), and differences in leaving group ability are reflected in changes in this quantity (ΔΔG‡). The quantity pKaH, however, represents the difference in energy between starting materials and products (ΔG) with differences in acidity reflected in changes in this quantity (ΔΔG). Also, the starting materials in these cases are different. In the case of pKa, the "leaving group" is bound to a proton in the starting material, while in the case of leaving group ability, the leaving group is bound to (usually) carbon. It is with these important caveats in mind that one must consider pKaH to be reflective of leaving group ability, but nevertheless the trends in each tend to correlate well with each other. Consistent with this picture, strong bases such as OH−, OR− and NR2− tend to make poor leaving groups, due their inability to stabilize a negative charge.
| Leaving groups ordered approximately in decreasing ability to leave [3] | |
|---|---|
| R-N2+ | dinitrogen |
| R-OR'2+ | dialkyl ether |
| R-OSO2RF | perfluoroalkylsulfonates (e.g. triflate) |
| R-OTs, R-OMs, etc. | tosylates, mesylates, and similar |
| R-I | iodide |
| R-Br | bromide |
| R-OH2+, R'-OHR+ | water, alcohols |
| R-Cl | chloride |
| R-ONO2, R-OPO(OH)2 | nitrate, phosphate, and other inorganic esters |
| R-SR'2+ | thioether |
| R-NR'3+, R-NH3+ | amines, ammonia |
| R-F | fluoride |
| R-OCOR | carboxylate |
| R-OAr | phenoxides |
| R-OH, R-OR | hydroxide, alkoxides |
| R-NR2 | amides |
It is exceedingly rare for groups such as H− (hydrides) and R3C− (alkyl anions, R=alkyl or H) to depart with a pair of electrons because of the instability of these bases.
Contextual differences in leaving group ability
It is important to note that the list given above is qualitative and describes trends. The ability of a group to leave is contextual. For example, in SNAr reactions, the rate is generally increased when the leaving group is fluoride relative to the other halogens. This effect is due to the fact that the highest energy transition state for this two step addition-elimination process occurs in the first step, where fluoride's greater electron withdrawing capability relative to the other halides stabilizes the developing negative charge on the aromatic ring. The departure of the leaving group takes place quickly from this high energy Meisenheimer complex, and since the departure is not involved in the rate limiting step, it does not affect the overall rate of the reaction. This effect is general to conjugate base eliminations.
Even when the departure of the leaving group is involved in the rate limiting step of a reaction there can still exist contextual differences that can change the order of leaving group ability. In Friedel-Crafts alkylations, the normal halogen leaving group order is reversed so that the rate of the reaction follows RF > RCl > RBr > RI. This effect is due to their greater ability to complex the Lewis acid catalyst, and the actual group that leaves is an "ate" complex between the Lewis acid and the departing leaving group.[4] This situation is broadly defined as leaving group activation
There can still exist contextual differences in leaving group ability in the purest form, that is when the actual group that leaves is not affected by the reaction conditions (by protonation or Lewis acid complexation) and the departure of the leaving group occurs in the rate determining step. In the situation where other variables are held constant (nature of the alkyl electrophile, solvent, etc.), a change in nucleophile can lead to a change in the order of reactivity for leaving groups. In the case below, tosylate is the best leaving group when ethoxide is the nucleophile, but iodide and even bromide become better leaving groups in the case of the thiolate nucleophile.[5]
| Leaving group (X) | 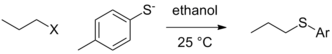 |  |
|---|---|---|
| Cl | 0.0074 | 0.0024 (at 40 °C) |
| Br | 1 | 1 |
| I | 3.5 | 1.9 |
| OTs | 0.44 | 3.6 |
Activation
It is common in E1 and SN1 reactions for a poor leaving group to be transformed into a good one by protonation or complexation with a Lewis acid. Thus, it is by protonation prior to departure that a molecule can formally lose such poor leaving groups as hydroxide.
The same principle is at work in the Friedel-Crafts reaction. Here, a strong Lewis acid is required to generate either a carbocation from an alkyl halide in the Friedel-Crafts alkylation reaction or an acylium ion from an acyl halide.
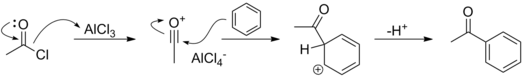
In the vast majority of cases, reactions that involve leaving group activation generate a cation in a separate step, prior to either nucleophilic attack or elimination. For example, SN1 and E1 reactions may involve an activation step, whereas SN2 and E2 reactions generally do not.
In conjugate base eliminations
The requirement for a good leaving group is relaxed in conjugate base elimination reactions. These reactions include loss of a leaving group in the β position of an enolate as well as the regeneration of a carbonyl group from the tetrahedral intermediate in nucleophilic acyl substitution. Under forcing conditions, even amides can be made to undergo basic hydrolysis, a process that involves the expulsion of an extremely poor leaving group, R2N−. Even more dramatic, decarboxylation of benzoate anions can occur by heating with copper or Cu2O, involving the loss of an aryl anion. This reaction is facilitated by the fact that the leaving group is most likely an arylcopper compound rather than the much more basic alkali metal salt.
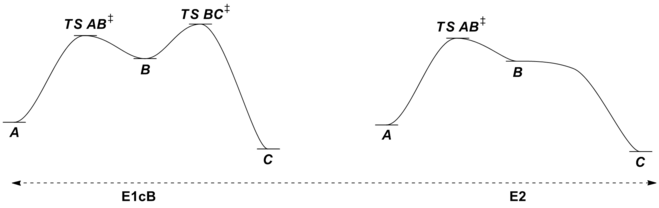
It should be noted that this dramatic departure from normal leaving group requirements occurs mostly in the realm of C=O double bond formation where formation of the very strong C=O double bond can drive otherwise unfavorable reactions forward. The requirement for a good leaving group is still relaxed in the case of C=C bond formation via E1cB mechanisms, but because of the relative weakness of the C=C double bond, the reaction still exhibits some leaving group sensitivity. Notably, changing the leaving group's identity (and willingness to leave) can change the nature of the mechanism in elimination reactions. With poor leaving groups, the E1cB mechanism is favored, but as the leaving group's ability changes, the reaction shifts from having a rate determining loss of leaving group from carbanionic intermediate B via TS BC‡ through having a rate determining deprotonation step via TS AB‡ (not pictured) to a concerted E2 elimination. In the latter situation, the leaving group X has become good enough that the former transition state connecting intermediates B and C has become lower in energy than B, which is no longer a stationary point on the potential energy surface for the reaction. Because only one transition state connects starting material A and product C, the reaction is now concerted (albeit very asynchronous in the pictured case) due to the increase in leaving group ability of X.
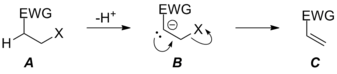
"Super" and "Hyper" leaving groups
The prototypical super leaving group is triflate, and the term has come to mean any leaving group of comparable ability. Compounds where loss of a super leaving group can generate a stable carbocation are usually highly reactive and unstable. Thus, the most commonly encountered organic triflates are methyl triflate and alkenyl or aryl triflates, all of which cannot form stable carbocations on ionization, rendering them relatively stable. It has been noted that steroidal alkyl nonaflates (another super leaving group) generated from alcohols and perfluorobutanesulfonyl fluoride were not isolable as such but immediately formed the products of either elimination or substitution by fluoride generated by the reagent. Mixed acyl-trifluoromethanesulfonyl anhydrides smoothly undergo Friedel-Crafts acylation without a catalyst,[6] unlike the corresponding acyl halides, which require a strong Lewis acid. Methyl triflate, however, does not participate in Friedel-Crafts alkylation reactions with electron-neutral aromatic rings.
Beyond super leaving groups in reactivity lie the "hyper" leaving groups. Prominent among these are λ3-iodanes, which include diaryl iodonium salts, and other halonium ions. In one study, a quantitative comparison of these and other leaving groups was conducted. Relative to chloride (krel=1), reactivities increased in the order bromide (krel=14), iodide (krel=91), tosylate (krel=3.7 x 104), triflate (krel=1.4 x 108), phenyliodonium tetrafluoroborate (PhI+ BF4−, krel=1.2 x 1014). Along with the criterion that a hyper leaving group be a stronger leaving group than triflate is the necessity that the leaving group undergo reductive elimination. In the case of halonium ions this involves reduction from a trivalent halonium to a monovalent halide coupled with the release of an anionic fragment. Part of the exceptional reactivity of compounds of hyper leaving groups has been ascribed to the entropic favorability of having one molecule split into three.

The ability of hyper leaving groups is enhanced by entropic factors
Dialkyl halonium ions have also been isolated and characterized for simple alkyl groups. These compounds, despite their extreme reactivity towards nucleophiles, can be obtained pure in the solid state with very weakly nucleophilic counterions such as SbF6−[7][8] and CHB11Cl11−.[9] The strongly electrophilic nature of these compounds engendered by their attachment to extremely labile R-X (R = alkyl, X = Cl, Br, I) leaving groups is illustrated by their propensity to alkylate very weak nucleophiles. Heating neat samples of (CH3)2Cl+ [CHB11Cl11]− under reduced pressure resulted in methylation of the very poorly nucleophilic carborane anion with concomitant expulsion of the CH3Cl leaving group. Dialkyl halonium hexafluoroantimonate salts alkylate excess alkyl halides to give exchanged products. Their strongly electrophilic nature, along with the instability of primary carbocations generated from ionization of their alkyl groups, points to their possible involvement in Friedel-Crafts alkylation chemistry.[7] The order of increasing lability of these leaving groups is R-I < R-Br < R-Cl.
See also
- Electrofuge
- Electrophile
- Elimination reaction
- Nucleofuge
- Nucleophile
- Substitution reaction
References
^ "Gold Book: leaving group" (PDF). IUPAC. doi:10.1351/goldbook.L03493..mw-parser-output cite.citation{font-style:inherit}.mw-parser-output q{quotes:"""""""'""'"}.mw-parser-output code.cs1-code{color:inherit;background:inherit;border:inherit;padding:inherit}.mw-parser-output .cs1-lock-free a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/6/65/Lock-green.svg/9px-Lock-green.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-limited a,.mw-parser-output .cs1-lock-registration a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/d/d6/Lock-gray-alt-2.svg/9px-Lock-gray-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-lock-subscription a{background:url("//upload.wikimedia.org/wikipedia/commons/thumb/a/aa/Lock-red-alt-2.svg/9px-Lock-red-alt-2.svg.png")no-repeat;background-position:right .1em center}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration{color:#555}.mw-parser-output .cs1-subscription span,.mw-parser-output .cs1-registration span{border-bottom:1px dotted;cursor:help}.mw-parser-output .cs1-hidden-error{display:none;font-size:100%}.mw-parser-output .cs1-visible-error{font-size:100%}.mw-parser-output .cs1-subscription,.mw-parser-output .cs1-registration,.mw-parser-output .cs1-format{font-size:95%}.mw-parser-output .cs1-kern-left,.mw-parser-output .cs1-kern-wl-left{padding-left:0.2em}.mw-parser-output .cs1-kern-right,.mw-parser-output .cs1-kern-wl-right{padding-right:0.2em}
^ "Gold Book: electrofuge" (PDF). IUPAC.
^ Smith, March. Advanced Organic Chemistry 6th ed. (501-502)
^ Brown, Herbert C.; Hans Jungk. "The Reaction of Benzene and Toluene with Methyl Bromide and Iodide in the Presence of Aluminum Bromide; Evidence for a Displacement Mechanism in the Methylation of Aromatic Compounds1,2". Journal of the American Chemical Society. 1955, 77 (21): 5584–5589. doi:10.1021/ja01626a039. ISSN 0002-7863.
^ Hoffmann, H. M. R. "1252. The rate of displacement of toluene-p-sulphonate relative to bromide ion. A new mechanistic criterion". Journal of the Chemical Society (Resumed). 1965: 6753–6761. doi:10.1039/JR9650006753. ISSN 0368-1769.
^ Martínez, A. Garcia; A. Herrera FernŽndez; D. Molero Vilchez; M. L. Laorden Gutiérrez; L. R. Subramanian. "A New Easy One-Step Synthesis of Isoquinoline Derivatives from Substituted Phenylacetic Esters". Synlett. 1993 (03): 229–230. doi:10.1055/s-1993-22413. ISSN 0936-5214.
^ ab Olah, George A.; John R. DeMember. "Friedel-Crafts chemistry. IV. Dialkylhalonium ions and their possible role in Friedel-Crafts reactions". Journal of the American Chemical Society. 1969, 91 (8): 2113–2115. doi:10.1021/ja01036a044. ISSN 0002-7863.
^ Olah, George A.; John R. DeMember. "Friedel-Crafts chemistry. V. Isolation, carbon-13 nuclear magnetic resonance, and laser Raman spectroscopic study of dimethylhalonium fluoroantimonates". Journal of the American Chemical Society. 1970, 92 (3): 718–720. doi:10.1021/ja00706a058. ISSN 0002-7863.
^ Stoyanov, Evgenii S.; Irina V. Stoyanova; Fook S. Tham; Christopher A. Reed (2010). "Dialkyl Chloronium Ions". Journal of the American Chemical Society. 132 (12): 4062–4063. doi:10.1021/ja100297b. ISSN 0002-7863. PMID 20218556.


